Artículos originales
Autoanticuerpos contra dianas neuronales en epilepsia
Autoantibodies against neuronal targets in epilepsy

 Viviana Novoa
Claudio Aranda
Viviana Novoa
Claudio Aranda
Autoanticuerpos contra dianas neuronales en epilepsia
Bioquímica y Patología Clínica, vol. 89, núm. 2, pp. 52-59, 2025
Asociación Bioquímica Argentina
Resumen: Introducción: La epilepsia autoinmune es poco reconocida y se desconoce su incidencia. Objetivo: Determinar la incidencia de autoanticuerpos neuronales en pacientes con epilepsia de etiología desconocida. Materiales y métodos: Se realizó un estudio observacional, longitudinal, prospectivo y analítico evaluando la presencia de autoanticuerpos para encefalitis autoinmune, ácido glutámico descarboxilasa-65 y onconeuronales en suero y líquido cefalorraquídeo, en pacientes consecutivos con epilepsia de etiología desconocida. Resultados: Se incluyeron 60 pacientes y 80 controles (30 sanos, 30 con esclerosis múltiple, 10 con lupus eritematoso sistémico, 10 con síndrome de Sjögren) para detectar anticuerpos neuronales. Un total de 28/60 (47%) pacientes con epilepsia tenía anticuerpos contra: receptor de N-metil-D-aspartato, proteína 2 asociada a contactina, proteína 1 inactivada de glioma rica en leucina y descarboxilasa del ácido glutámico, una incidencia que fue significativamente mayor (p < 0,001) que en la cohorte de control combinada. Pacientes y controles fueron negativos para anticuerpos onconeuronales, excepto 6 casos de epilepsia, 1 de esclerosis múltiple y 3 de lupus con a- descarboxilasa del ácido glutámico positivo por IIF-TBA e inmunotransferencia. No se encontró diferencia en la incidencia de los autoanticuerpos estudiados entre hombres y mujeres con epilepsia. La aparición de autoanticuerpos positivos en pacientes con epilepsias focales fue significativamente mayor en comparación con pacientes con epilepsia generalizada (p < 0,01). Conclusiones: Se han encontrado anticuerpos contra receptores (NMDAR), proteínas asociadas con VGKC (LGI1, CASPR2) y anticuerpos dirigidos a antígenos intracelulares (GAD65) en el suero y líquido cefalorraquídeo de pacientes con epilepsia, lo que sugiere una etiología autoinmune.
Palabras clave: epilepsia establecida, epilepsia de nuevo diagnóstico, autoanticuerpos neuronales, complejo de canales de potasio dependientes de voltaje, ácido glutámico descarboxilasa, receptor NMDA.
Abstract: Introduction: Autoimmune epilepsy is poorly recognized, and its incidence is unknown. Objective: To determine the incidence of neuronal autoantibodies in patients with epilepsy of unknown etiology. Materials and methods: An observational, longitudinal, prospective and analytical study was carried out to evaluate the presence of autoantibodies for autoimmune encephalitis, glutamic acid decarboxylase-65 and onconeuronal antibodies in serum and cerebrospinal fluid in consecutive patients with epilepsy of unknown etiology. Results: A total of 60 patients and 80 controls (30 healthy ones, 30 with multiple sclerosis, 10 with systemic lupus erythematosus, and 10 with Sjögren's syndrome) were included to detect neuronal antibodies. A total of 28/60 (47%) epilepsy patients had antibodies against N-methyl-D-aspartate receptor, contactin-associated protein 2, leucine-rich glioma-inactivated protein 1 and glutamic acid decarboxylase, an incidence that was significantly (p < 0.001) higher than that in the combined control cohort. Patients and controls were negative for onconeuronal antibodies, except six cases of epilepsy, one case of multiple sclerosis and three cases of Lupus with positive a-glutamic acid decarboxylase by indirect immunofluorescence tissue-based assay and immunoblotting. No difference was found in the incidence of the autoantibodies studied between male and female patients with epilepsy. The incidence of positive autoantibodies in patients with focal epilepsies was significantly higher than that in patients with generalized epilepsy (p < 0.01). Conclusions: Antibodies against receptors (NMDA receptor), proteins associated with VGKC (LGI1, CASPR2) and antibodies directed at intracellular antigens (GAD65) were found in the serum and cerebrospinal fluid of patients with epilepsy, suggesting an autoimmune etiology.
Keywords: established epilepsy, newly diagnosed epilepsy, neuronal autoantibodies, voltage-gated potassium channel complex, glutamic acid decarboxylase, NMDA receptor.
Introducción
Las crisis epilépticas son una de las presentaciones clínicas más frecuentes de la encefalitis autoinmune y, en algunos casos, puede ser el único síntoma. La incidencia de autoanticuerpos neuronales en la epilepsia de etiología desconocida varía entre los estudios. Se han identificado anticuerpos en pacientes con encefalitis límbica (EL)1 y más recientemente, en pacientes con convulsiones distónicas faciobraquiales (CDFB)2. Estas encefalopatías a menudo muestran un curso monofásico con títulos de anticuerpos que disminuyen sustancialmente en 1 a 2 años3. Dado que estos anticuerpos contra antígenos neuronales juegan un papel directo o indirecto en la transducción de señales sinápticas, la autoinmunidad asociada se manifiesta con convulsiones y síntomas neuropsiquiátricos. Las condiciones resultantes incluyen formas especiales de encefalitis límbica autoinmune, neuromiotonía o síndrome de Morvan. Estos síndromes graves y potencialmente letales pueden tener una etiología no paraneoplásica o paraneoplásica. Los síndromes neurológicos paraneoplásicos (SNP) son un conjunto de manifestaciones del sistema nervioso (SN), de etiopatogenia desconocida, que están relacionadas con la presencia de una neoplasia maligna y que no obedecen a invasión directa por el tumor, metástasis de este, trastornos metabólicos ni nutricionales, infecciones oportunistas ni son consecuencias de la radioterapia o quimioterapia para el tratamiento del cáncer. Los SNP son efectos indirectos del cáncer, en el que median mecanismos inmunológicos que permiten el diagnóstico mediante la detección de anticuerpos específicos, denominados anticuerpos onconeuronales (AON). Graus y col. definen los AON frente a antígenos intracelulares “como anticuerpos paraneoplásicos bien caracterizados que incluyen al anti–Hu o anticuerpos antinúcleo de las neuronas (ANNA-1), anti-Ri (ANNA-2), anti-Yo, anti-CV2/CRMP5, anti-Ma2, antianfifisina y anti-Tr (PCATr); y anticuerpos neurológicos paraneoplásicos parcialmente caracterizados, donde se encuentran los anti-Zic4, anti-SOX y también, los anti-ANNA-3 y PCA-2”.
La frecuencia de tumores subyacentes oscila entre el 10 y el 70 %, según el tipo de anticuerpo. Lo más probable es que estos anticuerpos desempeñen un papel causal en la patogenia. Dado que la terapia apropiada (intervención inmunomoduladora, resección del tumor) da como resultado una regresión considerable de los síntomas en la mayoría de los pacientes, el diagnóstico temprano es importante para un pronóstico favorable.
Los anticuerpos contra proteínas neuronales son comunes en pacientes que presentan convulsiones agudas de origen autoinmune sospechoso, según lo determinado por cambios inflamatorios en el líquido cefalorraquídeo o en neuroimágenes4. También se han informado anticuerpos contra las proteínas del complejo de canales de potasio dependientes de voltaje (VGKC) y la descarboxilasa del ácido glutámico (GAD) en una pequeña proporción de pacientes que presentaban convulsiones como síntoma principal o único y sin una implicación autoinmune manifiesta5. La mayoría de dichos informes han sido de naturaleza transversal, a menudo, reclutando pacientes de centros de referencia terciarios y, por lo tanto, sesgando a la población del estudio a favor de aquellos con una historia prolongada de epilepsia predominantemente refractaria. Por lo tanto, no está claro si la frecuencia elevada de autoanticuerpos contra antígenos neuronales observados en estos estudios es la causa subyacente de la epilepsia o simplemente una consecuencia de las convulsiones no controladas y cualquier daño neurológico asociado.
Recientemente, los anticuerpos contra otras proteínas expresadas en el cerebro, incluido el receptor de N-metil-D-aspartato (NMDAR), el receptor del ácido a-amino-3-hidroxi-5-metil-4-isoxazolpropiónico (AMPAR), el receptor B del ácido ᾲ-aminobutírico (GABABR) y los receptores de glicina (GLY-Rs), también se han informado en pacientes con encefalopatías6, pero la incidencia de estos nuevos anticuerpos sigue siendo relativamente desconocida en cohortes más grandes de epilepsia esporádica. Tampoco está claro si coexisten con anticuerpos informados más ampliamente, como VGKC y GAD, en un trastorno convulsivo con autoinmunidad amplia.
El objetivo de este estudio fue determinar la incidencia de varios autoanticuerpos neuronales nuevos y establecidos en una cohorte de pacientes con epilepsia.
Materiales y métodos
Se realizó un estudio observacional, longitudinal, prospectivo y analítico evaluando la presencia de autoanticuerpos para encefalitis autoinmune, ácido glutámico descarboxilasa-65 y onconeuronales en suero y líquido cefalorraquídeo en pacientes consecutivos con epilepsia establecida o epilepsia de inicio reciente de etiología desconocida.
Se excluyeron específicamente los sujetos con antecedentes de abuso de alcohol o drogas recreativas, sospecha de convulsiones no epilépticas o de un trastorno neurológico progresivo. Los análisis de laboratorio se realizaron por indicación del neurólogo de los pacientes como parte de las pruebas complementarias para realizar el diagnóstico diferencial ante sospecha de enfermedades neuroinmunológicas autoinmunes.
El suero y el líquido cefalorraquídeo (LCR) de los pacientes se analizaron mediante ensayo basado en tejido (IIF-TBA), ensayo basado en células transfectadas (CBA) e inmunotransferencia.
El IIF-TBA se realizó en un sustrato compuesto de cerebelo de primate. Se aplicaron las muestras de los pacientes (dilución de suero 1:10 y 1:100, dilución de LCR 1:1 y 1:5). Después de 30 minutos de incubación y lavado con buffer fosfato (PBS), el sustrato se incubó con anti-IgG-fluoresceína durante otros 30 minutos. Después de lavar con PBS, los portaobjetos fueron visualizados mediante microscopía de fluorescencia con aumento de 400x por dos operadores independientes.
Las muestras se analizaron con un kit de inmunotransferencia comercial (EUROLINE Paraneoplastic Neurological Syndromes 12 Ag; Euroimmun, Lübeck, Alemania) siguiendo las instrucciones del fabricante. Las tiras reactivas se escanearon y evaluaron para determinar la intensidad de la banda de Hu (ANNA1), Yo (PCA1), CV2 (CRMP5), Ri (ANNA2), Ma2, Tr (DNER), Zic-4, SOX1, recoverina, GAD, titina y anfifisina utilizando el software EUROLineScan (Euroimmun Lübeck, Alemania). Siguiendo las recomendaciones del fabricante, los valores de intensidad de banda de anticuerpos onconeurales ≤10 se consideraron negativos.
Los anticuerpos que reconocen antígenos de la superficie neuronal se determinaron por CBA (dilución 1:10 en suero; dilución 1:1 en LCR). Se investigaron los anticuerpos dirigidos contra los receptores de glutamato (tipo NMDA o tipo AMPA), receptores GABAb, dipeptidyl aminopeptidase-like protein 6 (DPPX), canales de potasio dependientes de voltaje (VGKC) o proteínas asociadas a VGKC (leucine-rich glioma-inactivated protein 1 [LGI1], contactin-associated protein 2 [CASPR2]), (Autoimmune Encephalitis Mosaic 1 Biochip 6, Euroimmun, Lübeck, Alemania).
Análisis estadístico
Los resultados de anticuerpos se expresaron como incidencia relativa en todo momento y se analizaron estadísticamente mediante la prueba de chi-cuadrado o la prueba exacta de Fisher, según correspondiera. Las comparaciones estadísticas de datos demográficos y características clínicas se realizaron mediante la prueba de Mann-Whitney (variables continuas) o la prueba de chi-cuadrado (variables categóricas), según correspondiera.
Consideraciones éticas
La información se obtuvo de una base de datos anonimizada para procurar la protección de los datos personales de los sujetos que participaron de la investigación.
Resultados
Entre enero de 2017 y marzo de 2023, analizamos 60 pacientes con epilepsia establecida o epilepsia de inicio reciente (suero, n = 60; LCR, n = 60). La mediana de edad fue de 48 años (rango 15-65), y 27 (45%) eran mujeres.
Los pacientes con epilepsia más 80 controles (30 voluntarios sanos, 30 con esclerosis múltiple [EM], 20 controles autoinmunes, 10 con lupus eritematoso sistémico, 10 con síndrome de Sjögren) se analizaron para detectar anticuerpos en suero y LCR contra NMDAR, AMPAR, CASPR2, LGI1, DPPX, GABAb y GAD. En los controles sanos se analizó solo el suero. En la Tabla I, se resumen los resultados.
Un total de 28/60 (47%) de pacientes con epilepsia tenía anticuerpos séricos contra NMDAR, CASPR2, LGI1 y GAD, una incidencia que fue significativamente (p < 0,001) mayor que en la cohorte de control combinada (Tabla I, Figura 2).
El anticuerpo más común fue a-NMDAR, encontrado en 15 pacientes con epilepsia (25 % del total) y solo en 4 de los controles (5 % del total). También se detectaron anticuerpos contra CASPR2, LGI1 y GAD (3,3 %, 8,3 % y 10 % de los pacientes con epilepsia, respectivamente) (Figura 1).
Todos los pacientes con epilepsia y los controles fueron negativos para anticuerpos onconeuronales, excepto 6 casos de epilepsia, 1 de EM y 3 de LES con a-GAD positivo por IIF-TBA e inmunotransferencia, (Figura 3).
No hubo diferencia en la incidencia de los autoanticuerpos estudiados entre hombres y mujeres con epilepsia (Tabla II). Se encontró una incidencia significativamente mayor de autoanticuerpos positivos en pacientes con epilepsias focales en comparación con pacientes con epilepsia generalizada (p < 0,01; Tabla II).
| n | a-NMDAR (CBA) | a-AMPAR (CBA) | a-CASPR2 (CBA) | a-LGI1 (CBA) | a-DPPX (CBA) | a-GABAb(CBA) | a-GAD (IIF-TBA-IT) | Total, Ac + | p | |
| Casos | ||||||||||
| Epilepsia | 60 | 15 | 0 | 2 | 5 | 0 | 0 | 6 | 28 | |
| Controles | ||||||||||
| Sanos | 30 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | <0,001* |
| EM | 30 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | <0,001* |
| Autoinmunes | 20 | 3 | 0 | 0 | 0 | 0 | 0 | 3 | 6 | <0,01* |
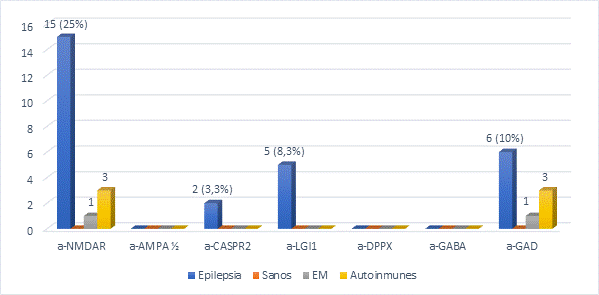
Figura 1.
Autoanticuerpos neuronales en casos y controles
Los pacientes con epilepsia (n = 60), los controles de enfermedad (esclerosis múltiple [EM]: n = 30; enfermedades autoinmunes: lupus eritematoso sistémico [LES] + síndrome de Sjögren [SS]: n = 20 y los controles sanos: n = 30) se analizaron buscando autoanticuerpos contra una variedad de proteínas neuronales. La figura muestra los resultados de la detección de anticuerpos anti- NMDAR, receptor de N-metil-D-aspartato; AMPAR, receptor del ácido amino-3-hidroxi-5-metil-4-isoxazolpropiónico; CASPR2, proteína asociada a contactina-2; LGI1, proteína inactivada-1 de glioma rico en leucina; DPPX, dipeptidyl-peptidase–like protein 6; GABAb, receptores acoplados a proteínas G para el ácido gamma-aminobutírico por ensayo basado en células transfectadas (CBA) y GAD, ácido glutámico descarboxilasa por inmunoblot e inmunofluorescencia por ensayo basado en tejido cerebelo de primate (IIF-TBA).
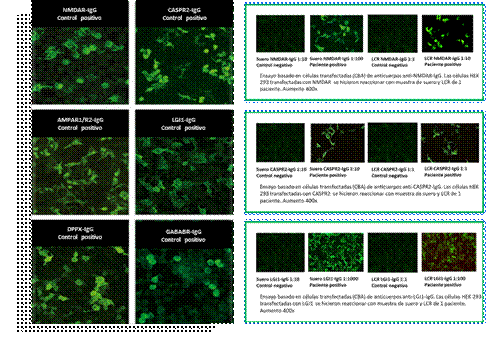
Figura 2.
Mosaico encefalitis autoinmune. Ensayo basado en células transfectadas
Determinación monoespecífica de autoanticuerpos dirigidos contra antígenos de la superficie neuronal por ensayo de inmunofluorescencia indirecta basado en células transfectadas que presentan los antígenos relevantes en su forma auténtica o conformacional. Los sustratos celulares transfectados de manera diferente se pueden combinar en multiplex. La figura de la izquierda muestra los controles positivos de la detección de anticuerpos anti- NMDAR, receptor de N-metil-D-aspartato; AMPAR, receptor del ácido amino-3-hidroxi-5-metil-4-isoxazolpropiónico; CASPR2, proteína asociada a contactina-2; LGI1, proteína inactivada-1 de glioma rico en leucina; DPPX, dipeptidyl-peptidase–like protein 6; GABAb, receptores acoplados a proteínas G para el ácido gamma-aminobutírico. A la derecha, se presentan las imágenes de inmunofluorescencia positiva de 3 pacientes.
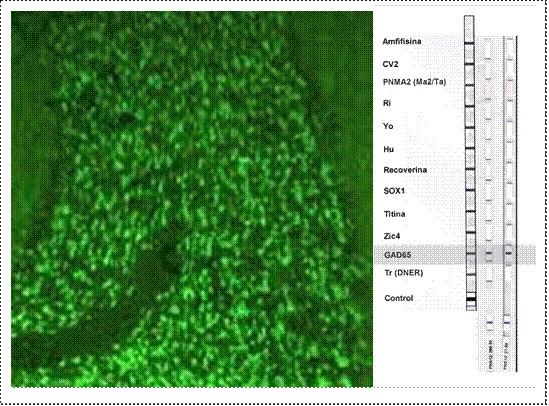
Figura 3.
Autoanticuerpos anti-ácido glutámico descarboxilasa
(a) Hallazgos de inmunofluorescencia en un caso de epilepsia asociada a anticuerpos anti-GAD (ácido glutámico descarboxilasa). Micrografía que muestra inmunofluorescencia utilizando suero del paciente diluido 1:100 en cerebelo (400x). El patrón de fluorescencia típico de los anticuerpos anti-GAD presenta una fluorescencia prominente en la capa de células granulares; a diferencia de los anti-anfifisina, no se tiñe la capa molecular. (b) Anti-GAD detectado por inmunoblot en suero y líquido cefalorraquídeo del mismo paciente. Dilución de suero 1:100, dilución de líquido cefalorraquídeo 1:2. CV2: CV2/ proteína mediadora de la respuesta a colapsina (CRMP)5; PNMA2/Ma2: familia del antígeno Ma paraneoplásico (PNMA), que está representada por al menos quince miembros de la familia; tres de los miembros de la familia, PNMA1-3, están asociados con el trastorno paraneoplásico; Ri: 2 proteínas con un peso molecular de 55 y 80 kDa, que se denominan Nova-1 y Nova-2. Su función parece estar relacionada con la regulación del preARNm; Yo: proteínas del citoplasma de las células de Purkinje del cerebelo de 34 y 62 kDa; Hu: familia de proteínas de unión al ARN (Hu-D, Hu-C y Hel N1), de tamaño molecular de 35-40 kDa, localizadas en los núcleos y el citoplasma de todas las neuronas y también, en las células tumorales; SOX1: antígeno localizado en los núcleos de las células gliales de Bergmann del cerebelo adulto; Zic4: proteínas de los núcleos de la capa granular, perteneciente a una familia de cinc-proteínas que tienen un papel importante en el desarrollo del sistema nervioso; GAD65: acido glutámico descarboxilasa, Tr (DNER): antígeno que se expresa tanto en el citoplasma de células de Purkinje como en axones y dendritas.
| n | a-NMDAR (CBA) | a-AMPAR (CBA) | a-CASPR2 (CBA) | a- LGI1 (CBA) | a- DPPX (CBA) | a-GABAb(CBA) | a-GAD (IIF-TBA-IT) | Total Ac + | p | |
| Sexo | ns | |||||||||
| Masculino | 33 | 8 (24%) | 0 (0%) | 1 (3%) | 3 (9%) | 0 (0%) | 0 (0%) | 3 (9%) | 15 (45%) | |
| Femenino | 27 | 7 (26%) | 0 (0%) | 1 (4%) | 2 (7%) | 0 (0%) | 0 (0%) | 3 (11%) | 13 (48%) | |
| Tipo epilepsia | ||||||||||
| Generalizada | 20 | 5 (25%) | 0 (0%) | 0 (0%) | 1 (5%) | 0 (0%) | 0 (0%) | 1 (5%) | 7 (35%) | <0,01* |
| Focal | 40 | 10 (25%) | 0 (0%) | 2 (5%) | 4 (10%) | 0 (0%) | 0 (0%) | 5 (12%) | 21 (52%) |
Discusión
Estudios previos han demostrado la presencia de autoanticuerpos contra NMDAR7 y GAD8 en cohortes transversales de pacientes con epilepsia, en síndromes epilépticos bien definidos y en pacientes con autoinmunidad neurológica preexistente conocida o sospechada4. Los anticuerpos contra CASPR29 y LGI110se han examinado recientemente en una población con epilepsia autoinmune y los anticuerpos contra NMDAR solo se han investigado en pequeños estudios pediátricos, centrados en encefalitis límbica y estado epiléptico11.
En este estudio, buscamos anticuerpos en una cohorte de pacientes consecutivos, no seleccionados, con epilepsia. Encontramos anticuerpos positivos para NMDAR, CASPR2, LGI1 y GAD con una incidencia significativamente mayor que en controles sanos o enfermos. En total, 28 (47%) de 60 pacientes con epilepsia, en este análisis, fueron positivos para anticuerpos contra antígenos neuronales.
Ninguno de los pacientes en este estudio tenía evidencia clínica de EL, encefalitis asociada a a-NMDAR o cualquier otra enfermedad neuroinflamatoria mediada por anticuerpos. De esta manera, nuestros hallazgos sugieren que varios autoanticuerpos neurológicos asociados con encefalopatías de inicio subagudo también pueden dar lugar a epilepsia esporádica, de forma semejante a lo descripto en informes previos en una cohorte de pacientes jóvenes con epilepsia de inicio inexplicable12. Los seis pacientes con anticuerpos anti-GAD, 3 mujeres y 3 hombres, tenían epilepsia focal, lo que es consistente con otros estudios13 e informes más recientes de una relación entre anti-GAD y epilepsias que surgen en el lóbulo temporal14,15. De manera similar, los anticuerpos anti-GAD también se informaron recientemente en una forma de EL en mujeres adultas jóvenes con epilepsia del lóbulo temporal y trastornos cognitivos leves16. Después de a-NMDAR, a-GAD fue el autoanticuerpo más frecuente en nuestro análisis. Esto no es extraño, ya que, a diferencia de la mayoría de los autoanticuerpos neuronales, los anticuerpos a-GAD se han asociado durante muchos años con epilepsias resistentes a los medicamentos sin cumplir los criterios de encefalitis límbica17, lo cual es congruente con los criterios de selección de nuestro estudio.
Siete (11,6 %) de 60 pacientes con diagnóstico de epilepsia focal y generalizada, en nuestro estudio, tenían autoanticuerpos a-CASPR2 (2/60 3,3 %) y a-LGI1 (5/60 8,3%), lo cual es consistente con la prevalencia relativa de estos anticuerpos en otros estudios. Los pacientes con epilepsias generalizadas, generalmente, tienen una base genética para sus convulsiones, aunque, en la mayoría de los casos. esto no ha sido confirmado formalmente. Este tema se aborda en la revisión de la terminología y los conceptos para la organización de las convulsiones y las epilepsias que realizó la Liga Internacional contra la Epilepsia (ILAE), que se revisó en 2017, la cual identificó como "Inmune" una de las etiologías de la epilepsia por primera vez18. Los autoanticuerpos dirigidos contra los mismos canales iónicos y receptores a menudo implicados en las epilepsias genéticas podrían teóricamente dar lugar a un fenotipo idéntico. Otros estudios sobre cohortes más grandes de pacientes con epilepsia generalizada para explorar esta posibilidad son así justificados, y futuras revisiones a la clasificación de epilepsias deben considerar la autoinmunidad como un contribuyente etiológico.
La encefalitis anti-NMDAR es la enfermedad más frecuente entre los EA con anticuerpos anti-NSA19. Un estudio prospectivo, multicéntrico y basado en la población sugiere que los trastornos autoinmunes son la tercera causa más común de encefalitis, después de las infecciones, generalmente virales, y la encefalomielitis diseminada aguda, que suele ser un trastorno posinfeccioso20. El diagnóstico de encefalitis anti-NMDAR se confirma con la detección de anticuerpos en LCR contra la subunidad GluN1 de NMDAR; las pruebas de suero son menos fiables, puesto que dan como resultados falsos negativos hasta en el 14 % de los casos21. En pacientes con títulos bajos de anticuerpos, los anticuerpos séricos se vuelven indetectables después de la fase aguda, pero los anticuerpos en el líquido cefalorraquídeo no desaparecen en un corto período de tiempo. Por otro lado, los casos con suero positivo/líquido cefalorraquídeo negativo deben considerarse falsos positivos y deben reevaluarse.
El epítopo antigénico de este anticuerpo es NR1 (GluN1), y uno de los dominios extracelulares de la subunidad de GluN1 es la región N368/G369, que es crucial para la creación de inmunorreactividad,; su autoanticuerpo se denomina anticuerpo anti-GluN1 o anti-NMDAR. Sin embargo, este anticuerpo no identifica la estructura primaria o epítopo lineal en GluN1, ya que es un autoanticuerpo que reconoce el epítopo conformacional del receptor22,23. Este anticuerpo no interfiere con la activación mediada por el complemento, sino que promueve la internalización de NMDAR, lo que reduce la cantidad de NMDAR expresados y la función de NMDAR como una red completa24.
El anticuerpo anti-LGI1 fue previamente llamado anti-VGKC. El anticuerpo anti-VGKC ha sido medido por el método de radioinmunoanálisis (RIA), sin embargo, el verdadero epítopo antigénico no fue el propio VGKC, sino LGI1 y CASPR2. LGI1 es una proteína intersináptica que se une a los factores de adhesión celular presinápticos y postsinápticos ADAM23 y ADAM22, y estabiliza AMPAR en la membrana postsináptica. Por otro lado, CASPR2 se expresa tanto en nervios centrales como periféricos, y se detectan anticuerpos anti-CASPR2 en el síndrome de Isaacs y el síndrome de Morvan. Con base en estos resultados, los anticuerpos anti-VGKC pasaron a llamarse anticuerpos del complejo anti-VGKC, pero existe la opinión de que se debe usar el verdadero nombre del antígeno en lugar del complejo VGKC25. Posteriormente, 90 de 162 pacientes (56%) en quienes se detectaron anticuerpos anti-complejo VGKC en suero reconocieron LGI1 o CASPR2, mientras que el 38% restante (27/72) reconoció Kv1; se reportó que el 59% (16 /27) de ellos reconoció el componente intracelular de Kv1. Por lo tanto, los anticuerpos del complejo anti-VGKC dobles seronegativos ya no pueden considerarse anticuerpos anti-antígenos neurales de superficie (anti-NSA) y, en los casos de doble seronegatividad, los resultados de anticuerpos medidos por el método RIA no deben utilizarse como base para iniciar la inmunoterapia por lo que se concluye que los anticuerpos deben medirse mediante CBA26.
La incidencia de cada anticuerpo obtenida en nuestro análisis puede guiar la investigación en términos de qué autoanticuerpos se pueden determinar en estudios futuros, como así también, en términos clínicos. La heterogeneidad en el tipo de anticuerpo y en el método para su determinación es alta entre los estudios. Se necesitan más estudios con protocolos homogéneos de pruebas de autoanticuerpos neuronales para obtener un resultado más preciso de la incidencia real de autoanticuerpos neuronales y también para encontrar otros parámetros a fin de mejorar la comprensión actual de la importancia clínica de los autoanticuerpos neuronales en pacientes con epilepsia de origen desconocido.
En esta serie prospectiva, consecutiva de pacientes con epilepsia, un número significativo tenía niveles detectables de anticuerpos neurológicos en suero y LCR, lo que sugiere una etiología autoinmune. Este estudio proporciona una buena estimación de la incidencia de estos anticuerpos y sugiere que la epilepsia autoinmune es poco reconocida y no es infrecuente.
Nuestro estudio tuvo limitaciones debido a que la prueba para anticuerpos anti receptor de glicina (a-GlyR) no estaba disponible en el laboratorio y porque algunos pacientes con epilepsia autoinmune pueden tener un autoanticuerpo neurológico todavía no reconocido, así que los resultados aún pueden representar una subestimación de la incidencia de la epilepsia autoinmune.
Conclusiones
Según nuestros resultados, podemos concluir que anticuerpos contra receptores (NMDAR), proteínas asociadas con VGKC (LGI1, CASPR2) y anticuerpos dirigidos a antígenos intracelulares (GAD65) se encuentran en el suero y líquido cefalorraquídeo de pacientes con epilepsia. Estos pacientes en los que se han detectado anticuerpos frente a proteínas neuronales clínicamente relevantes pueden responder muy bien a la terapia inmunomoduladora.
Los futuros estudios de detección neuroinmunológica deben tratar de incluir preferentemente cohortes prospectivas de pacientes con epilepsia nueva o de inicio reciente para comprender mejor la contribución de los autoanticuerpos a la fisiopatología de la epilepsia, para promover un diagnóstico rápido y preciso y para fomentar la consideración de opciones de tratamiento alternativas, incluyendo el posible uso de inmunoterapias.
Referencias bibliográficas
1. Vincent A, Buckley C, Schott JM, Baker I, Dewar BK, Detert N, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004; 127:701-712, https://doi.org/10.1093/brain/awh077
2. Irani SR, Michell AW, Lang B, Pettingill P, Waters P, Johnson MR, et al. Fachiobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011;69:892-900, https://doi.org/10.1002/ana.22307
3. Buckley C, Oger J, Clover L, T€uz€un-E, Carpenter K, Jackson M, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 2001;50:73-78, https://doi.org/10.1002/ana.1097
4. Quek AML, Britton JW, McKeon A, So E, Lennon VA, Shin C, et al. Autoimmune epilepsy: clinical characteristics and response to immunotherapy. Arch Neurol 2012;69:582-593, https://doi.org/10.1001/archneurol.2011.2985
5. Kwan P, Sills GJ, Kelly K, Butler E, Brodie MJ. Glutamic acid decarboxylase autoantibodies in controlled and uncontrolled epilepsy: a pilot study. Epilepsy Res 2000;42:191-195, https://doi.org/10.1016/s0920-1211(00)00180-7
6. Vincent A, Crino PB. Systemic and neurologic autoimmune disorders associated with seizures or epilepsy. Epilepsia. 2011;52:12-17, https://doi.org/10.1111/j.1528-1167.2011.03030.x
7. Chena Z, Zhoua J, Wu D, Ji C, Luo B, Wang K. Altered executive control network connectivity in anti-NMDA receptor encephalitis. Ann Clin Trans Neurol. 2022;9(1):30-40, https://doi.org/10.1002/acn3.51487
8. Tröscher A, Mair K, Verdú de Juan L, Köck U, Steinmaurer A, Baier H, et al. Temporal lobe epilepsy with GAD antibodies: neurons killed by T cells not by complement membrane attack complex. Brain. 2023;146:1436-1452, https://doi.org/10.1093/brain/awac404
9. Garrido-Sanabria E, Zahid A, Britton J, Kraus G, López-Chiriboga AS, Zekeridou A, et al. CASPR2-IgG-associated autoimmune seizures. Epilepsia. 2022;63(3):709-722, https://doi.org/10.1111/epi.17164
10. Smith KM, Dubey B, Liebo GB, Flanagan EP, Britton J. Clinical Course and Features of Seizures Associated with LGI1-Antibody Encephalitis. Neurology. 2021;97(11):e1141-e1149, https://doi.org/10.1212/wnl.0000000000012465
11. Haberlandt E, Bast T, Ebner A, Holthausen H, Kluger G, Kravljanac R, et al. Limbic encephalitis in children and adolescents. Arch Dis Child. 2011;96:186-191, https://doi.org/10.1136/adc.2010.183897
12. Niehusmann P, Dalmau J, Rudlowski C, Vincent A, Elger CE, Rossi JE, et al. Diagnostic value of N-methyl-D-aspartate receptor antibodies in women with new-onset epilepsy. Arch Neurol 2009;66(4):458-464, https://doi.org/10.1001/archneurol.2009.5
13. Irani S, Bien C, Lang B. Autoimmune epilepsies. Curr Opin Neurol 2011;24:146-153, https://doi.org/10.1097/wco.0b013e3283446f05
14. Liimatainen S, Peltola M, Sabater L, Fallah M, Kharazmi E, Haapala AM, et al. Clinical significance of glutamic acid decarboxylase antibodies in patients with epilepsy. Epilepsia 2010;51:760-767, https://doi.org/10.1111/j.1528-1167.2009.02325.x
15. Lin Nan, Bai Lin, Liu Qing, Chen Jianhua, Ren Haitao, Guan Hongzhi et al. Seizure semiology and predictors of outcomes in Chinese patients with glutamic acid decarboxylase antibody-associated neurological syndrome. BMC Neurol 2023;23(1): 149, https://doi.org/10.1186/s12883-023-03182-x
16. Balagopal K, Shenoy SG, Dutta D, et al. Autoimmune limbic encephalitis associated with glutamic acid decarboxylase antibodies. J Evolution Med Dent Sci 2021;10(28):2131-2133, https://doi.org/10.14260/jemds/2021/435
17. Graus F, Saiz A, Dalmau J. GAD antibodies in neurological disorders-Insights and challenges. Nat Rev Neurol. 2020;16:353-365, https://doi.org/10.1038/s41582-020-0359-x
18. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia 2017;58(4):512-521, https://doi.org/10.1111/epi.13709
19. Dalmau J, Graus F. Antibody-mediated encephalitis. N Engl J Med 2018; 378(9):840-851, https://doi.org/10.1056/nejmra1708712
20. Granerod J, Ambrose HE, Davies NW, et al. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis 2010;10(12):835-44, https://doi.org/10.1016/s1473-3099(10)70222-x
21. Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol 2014;13(2):167-77, https://doi.org/10.1016/s1474-4422(13)70282-5
22. Gleichman AJ, Spruce LA, Dalmau J, et al. Anti-NMDA receptor encephalitis antibody binding Is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J Neurosci 2012;32(32):11082-94, https://doi.org/10.1523/jneurosci.0064-12.2012
23. Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol 2014;13(2):167-177, https://doi.org/10.1016/s1474-4422(13)70282-5
24. Dalmau J, Lancaster E, Martinez-Hernandez E, et al. Clinical experience and laboratory investigations in patients with antiNMDAR encephalitis. Lancet Neurol 2011;10(1):63-74, https://doi.org/10.1016/s1474-4422(10)70253-2
25. van Sonderen A, Schreurs MW, Wirtz PW, et al. From VGKC to LGI1 and Caspr2 encephalitis: the evolution of a disease entity over time. Autoimmun Rev 2016; 15(10):970-974, https://doi.org/10.1016/j.autrev.2016.07.018
26. Lang B, Makuch M, Moloney T, et al. Intracellular and nonneuronal targets of voltage-gated potassium channel complex antibodies. J Neurol Neurosurg Psychiatry 2017;88(4):353-361, https://doi.org/10.1136/jnnp-2016-314758
Notas
Notas de autor
sgramos58@hotmail.com
Información adicional
redalyc-journal-id: 651